(UPDATE: A slightly updated version of this blog is now published here in PLoS Computational Biology)
(written by Paul Medvedev and Mihai Pop)
When you learned about genome assembly algorithms, you might have heard a story that goes something like this:
In the overlap-layout paradigm, solving the assembly problem requires solving the Hamiltonian cycle problem in the overlap graph. This is difficult, because the Hamiltonian cycle program is NP-hard. On the other hand, if we break our reads up into k-mers, we can build the de Bruijn graph. Then, the assembly problem becomes the problem of finding an Eulerian cycle in the de Bruijn graph, which is easily solvable in linear time. Thus, by formulating the assembly problem in terms of the de Bruijn graph, we can solve the much easier Eulerian cycle problem and not have to solve the NP-hard Hamiltonian cycle problem.
In this post, we explain that while de Bruijn graphs have indeed been very useful, the reason has nothing to do with the complexity of the Hamiltonian and Eulerian cycle problems.
Every Eulerian cycle in a de Bruijn graph or a Hamiltonian cycle in an overlap graph corresponds to a single genome reconstruction where all the repeats are completely resolved. For example, Figure 1 shows two different Eulerian cycles in the same graph (a similar example could be constructed for Hamiltonian cycles in an overlap graph). Each cycle corresponds to a different arrangement of segments between the repeats. The presence of multiple Eulerian or Hamiltonian cycles implies that the genome structure is ambiguous given the data available. In other words, using the same set of reads one can reconstruct different genomes, each of which is fully consistent with the data (Figure 1 gives an example). Choosing one of these reconstructions arbitrarily would be foolhardy since only one of them is the original genome. No sane assembly algorithm would do this, and that is one of the major reasons why an algorithm for finding Eulerian or Hamiltonian cycles is not part of any assembly algorithm used in practice.
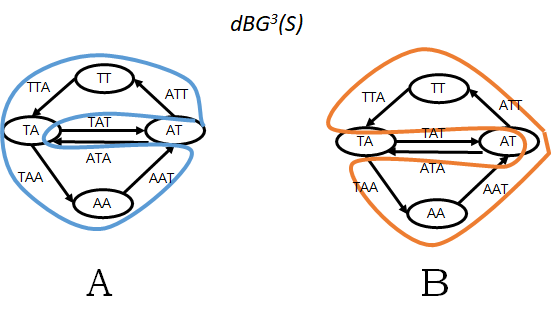
Figure 1: A worked out example for a set of reads R = {TATTA, TAATA} and k=3. Here, the set of all k-mers is S = spk(R) = {TAT, ATT, TTA, TAA, AAT, ATA}. Panel A shows G1 = dBGk(S) and one possible Eulerian cycle of G1 (in blue). Panel B show the only other Eulerian cycle in G1 (in orange). The genome reconstruction corresponding to the blue cycle is ATTAATAT and to the orange cycle is ATTATAAT (note that because the genome is circular, the last two characters of each string are equal to the first two characters).
Instead, assemblers output contigs—long, contiguous segments which can unambiguously be inferred to be part of the genome. Finding such segments is a very different computational problem than finding a single Eulerian or Hamiltonian cycle1 . In fact, it was shown that finding all possible contigs can be done in polynomial time, regardless of whether the genome reconstruction is modeled as a Hamiltonian or Eulerian cycle (Tomescu and Medvedev, 2016). The algorithm used in practice (the unitig algorithm) is linear and nearly identical in the two graph models.
Perhaps you are not convinced by the above reasoning? Fine. For the sake of argument, let’s imagine that we really are interested in finding a single, arbitrary, genome reconstruction. But even in this case, the distinction between Eulerian and Hamiltonian cycles is misleading. We make our point with this Theorem, which we first state informally (a formal statement and proof will come below):
Main Theorem (informal): The following problems are equivalent and solvable in linear time:
- Find an Eulerian cycle in the de Bruijn graph where the edges correspond to k-mers in the reads.
- Find a Hamiltonian cycle in the de Bruijn graph where the edges correspond to all the possible (k+1)-mers that can be obtained from the reads’ k-mers.
The first part of the theorem should not be surprising. It states one half of the story we started with, namely that we can solve the assembly problem in linear time by finding an Eulerian cycle in a de Bruijn graph. The second part of the theorem, though, adds a twist. It is about finding a Hamiltonian cycle, but it differs from the initial story in two ways. First, it is a Hamiltonian cycle in a de Bruijn graph, not in an overlap graph. This might seem strange, but there is no special connection between overlap graphs and the Hamiltonian cycle problem — one is free to find a Hamiltonian cycle in any graph they wish. Second, the problem is solvable in linear time in this case, even though it is NP-hard in general. This might also seem strange, but in fact it is common for NP-hard problems to have polynomial-time solutions for a restricted class of inputs2.
What the theorem states, then, is that one can solve the assembly problem in linear time by finding a Hamiltonian cycle within an appropriately defined de Bruijn graph. The fact that the Hamiltonian cycle problem is NP-hard in general graphs is not directly relevant. What is important is the underlying structure of the de Bruijn graph which makes the Hamiltonian cycle problem easy to solve3. Hence, the initial story was right in the sense that using de Bruijn graphs is a good idea but wrong to imply that the complexity of the Hamiltonian cycle problem is a reason. All of this is of course assuming we are, for some reason, interested in an arbitrary genome reconstruction, which, as we argued earlier, we typically are not.
So why are de Bruijn graphs so popular for short read assembly, if not for the difference in the complexity of finding Eulerian or Hamiltonian cycles? The answer is complex, which might explain why the initial simple story was appealing. It may have to do with the simplicity of their implementation, the appeal of the k-mer abstraction, the ease of error correction, or with something else. In fact, the difference between using de Bruijn graphs and overlap graphs is poorly understood and is a fascinating open research problem. But, the Eulerian and Hamiltonian cycle dichotomy is not really relevant to assembly or to the popularity of de Bruijn graphs.
Acknowledgements
PM would like to thank Rayan Chikhi for feedback on the post and Alexandru Tomescu and Michael Brudno for many helpful discussions on this topic (and Michael Brudno specifically for introducing him to the problem a long time ago).
References
- Bresler, Guy, Ma’ayan Bresler, and David Tse. “Optimal assembly for high throughput shotgun sequencing.” In BMC bioinformatics, vol. 14, no. S5, p. S18. BioMed Central, 2013.
- Carl Kingsford, Michael C. Schatz, and Mihai Pop. “Assembly complexity of prokaryotic genomes using short reads.” In BMC bioinformatics 11(1): 21, 2010.
- Medvedev, Paul, Konstantinos Georgiou, Gene Myers, and Michael Brudno. “Computability of models for sequence assembly.” In International Workshop on Algorithms in Bioinformatics, pp. 289-301. Springer, Berlin, Heidelberg, 2007.
- Medvedev, Paul. “Modeling biological problems in computer science: a case study in genome assembly.” Briefings in bioinformatics 20, no. 4 (2019): 1376-1383.
- Nagarajan, Niranjan, and Mihai Pop. “Parametric complexity of sequence assembly: theory and applications to next generation sequencing.” Journal of computational biology 16, no. 7 (2009): 897-908.
- Tomescu, Alexandru I., and Paul Medvedev. “Safe and complete contig assembly through omnitigs.” Journal of Computational Biology 24, no. 6 (2017): 590-602.
Proof of theorem
Let’s prove the main theorem, which follows almost directly from definitions. It may have been observed in previous papers, though we are not sure if it has been explicitly stated.
Let t be a string, k be a positive integer, and S be a set of k-mers. Let R be a set of reads, i.e. strings of length k. We define prei (t) and sufi (t) as the prefix and suffix, respectively, of length i of t. The k-spectrum of R, denoted by spk (R), is the set of all k-mer substrings of the strings of R. The de Bruijn graph of order k of S, denoted as dBGk(S), is defined as follows4. The vertex set is spk-1 (S) , and for every k-mer x ∈S, we add an edge from prek-1 (x) to sufk-1 (x) . Figure 1 shows an example of a de Bruijn graph. We define the closure of S, denoted closure(S), to be the set of all (k+1)-mers y such that prek(y) ∈ S and sufk(y) ∈ S. Informally, it is the set of all (k+1)-mers that can be constructed from S.
Main Theorem (formal): Let R be a set of strings whose smallest length is L. Let k be a positive integer less than L. Then, there is a one-to-one correspondence between Eulerian cycles in dBGk(spk(R) and Hamiltonian cycles in dbGk+1(closure(spk(R))). Moreover, an Eulerian or Hamiltonian cycle can be found in its respective graph in O(|spk(R)|) time.
Proof: Let S=spk(R). Let G1 = dBGk(S) and G2 = dBGk+1 (closure(S)). First, we show that the vertex set of G2 is S, i.e. spk(closure(S)) = S. Clearly, spk(closure(S)) ⊆ S, since no new k-mers are created during the closure process. Now, let x be a k-mer in S. It must appear in some read r, and since the length of r is greater than k, x must be a prefix or suffix of some (k+1)-mer y in R. Moreover, y must be in closure(S), since its prefix and suffix are both in r and hence in S. Therefore, x ∈ spk(closure(S), completing our proof that S ⊆ spk(closure(S)) and the vertex set of G2 is S.
Observe that a sequence of k-mers C1 = x0, …, xn-1 is a sequence of edges defining an Eulerian cycle in G1 if and only if the set of k-mers of C1 is exactly S (without any repetitions) and, for all i, sufk-1 (xi) = prek-1 (xi + 1 mod n). Also, observe that a sequence of k-mers C2 = x0, …, xn-1 is a sequence of vertices defining a Hamiltonian cycle in G2 if and only if the exact same criteria holds, i.e. the set of k-mers of C2 is exactly S (without repetitions) and, for all i, sufk-1 (xi) = prek-1 (xi+1 mod n). Thus, there is a one-to-one correspondence between Eulerian cycles in dBGk(S) and Hamiltonian cycles in dBGk+1(closure(S)). Figure 2 shows an example.
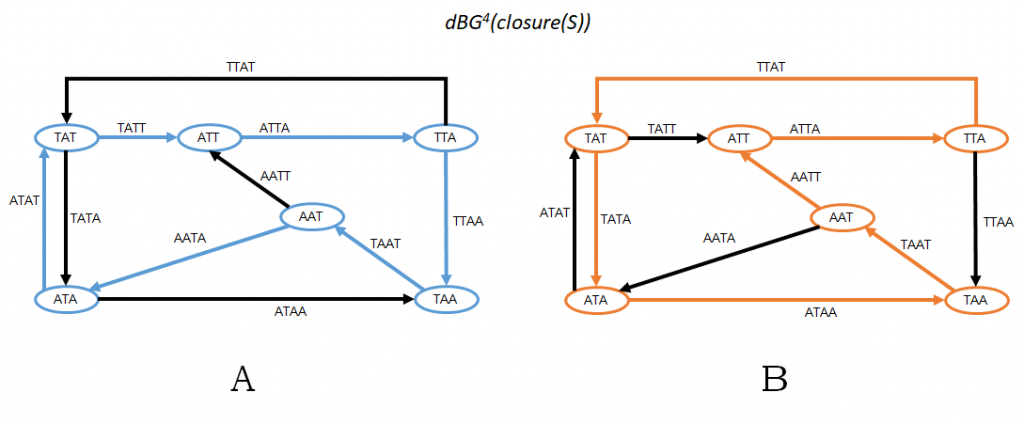
Figure 2: Using the same example as in Figure 1, this figure shows G2 = dBGk+1(closure(S)) and the two possible Hamiltonian cycles in G2. Notice that the k-mer sequence corresponding to the blue cycle in Panel A is the same as in Fig 1A; similarly, the k-mer sequence for the orange cycle in Panel B is the same as in Fig 1B.
For the running time, an Eulerian cycle can be found in time linear in the number of edges using a classical algorithm, e.g., Hierholzer’s Algorithm. Giving the one-to-one correspondence above, the vertex labels of a Hamiltonian cycle in G2 can be found by outputting the edge labels of an Eulerian cycle in G1. Hence, the running times for the two problems are equivalent.
∎
- For those who are curious, the problem of finding all possible contigs was first mentioned in Nagarajan and Pop (2009) and later explored by Tomescu and Medvedev (2016) and other follow-up papers. In this line of research, the set of substrings which must appear in all possible genome reconstructions are called omnitigs. For a broader discussion on how to formulate the genome assembly problem, see the tutorial by Medvedev (2019).
- For example, the satisfiability problem is NP-hard in general but is polynomial-time solvable when the clauses are restricted to have only two variables.
- There is much more that has been said on the complexity of finding a single genome reconstruction. For a starting point, see the papers by Medvedev and Brudno (2007), Nagarajan and Pop (2009), Kingsford, Schatz, and Pop (2010), and Bresler et al (2013). The last two papers especially show how the complexity depends more on the repeat structure of the underlying genome than on the graph model used.
- This graph is sometimes referred to as the edge centric dBG, not to be confused with a node centric one (see https://www.biostars.org/p/175058/#256741 for an explanation of the two terms).
Thanks for the blog! Although it is not practical to report a Hamiltonian cycle in either de Bruijn graphs or overlap graphs as assembly output for the reasons mentioned above, I’m curious to know whether the complexity of computing a Hamiltonian cycle in overlap graphs is known. Are overlap graphs (like DBGs) tractable input instances for the Hamiltonian cycle problem?
It depends on how you define an overlap graph. Braga and Meidanis (https://doi.org/10.1007/3-540-45995-2_10) show that for any directed graph, we can assign strings to the vertices so that there is an edge if and only if the strings overlap. Thus the set of overlap graphs is the set of all directed graphs, and so Hamiltonian cycle remains hard.
But if your overlap graph is a node centric dBG (we didn’t use this term in the theorem above, but this is what the second bullet point is about), then our theorem states that HC is solvable in poly time.
You might consider some intermediate definition of an overlap graph, but I’m not sure we know much in that case. The construction of Braga and Meidanis assigns strings whose length is exponential in the number of strings, which is not something that would happen in practice. We worked on this direction in the paper “On the readability of overlap digraphs”
(https://arxiv.org/abs/1504.04616), where a the “readability” of a graph is the smallest r such that we can assign strings of length r to each vertex so that the edges correspond to overlaps. Your question can then be restated as: is HC NP-hard on graphs of readability, say, o(n)? I don’t believe anything is published about this.
This makes sense. Thanks for referring those two papers, I was unaware of these results. Regarding your suggestion on restating the question in the above manner, I’m not sure if it would be fair to assume that read lengths are much smaller than count of reads because ONT has already made it feasible to sequence reads of length >1 Mb.
Learn a lot from this great article. Didn’t know Hamiltonian cycle in de Buijn graph is in P. Just a bit input, de Bruijn graph works great for short reads also because k-mer decomposition meets Illumina error-prone profile in high GC regions, where OLP hardly assemble through.
Thanks for the comment! Just to clarify, we didn’t show that HC on de Bruijn graphs is in P…our Theorem gives a linear time algorithm only for a subclass of de Bruijn graphs, the ones where the edges correspond to all the possible (k+1)-mers that can be obtained from the reads’ k-mers. These are sometimes called node centric de Bruijn graphs. If the edges correspond to an arbitrary set of (k+1)-mers, which they do in edge-centric de Bruijn graphs, then I don’t know of a poly-time HC algorithm.